
Imagine releasing a batch of life-saving drugs and you do not want to take any chance with the quality of the product. In pharmaceuticals, method validation isn’t just a checkbox but it’s the shield protecting patients and your company’s reputation. Compliance with FDA-like regulatory bodies’ analytical method validation is highly important.
What is method validation in Pharma?
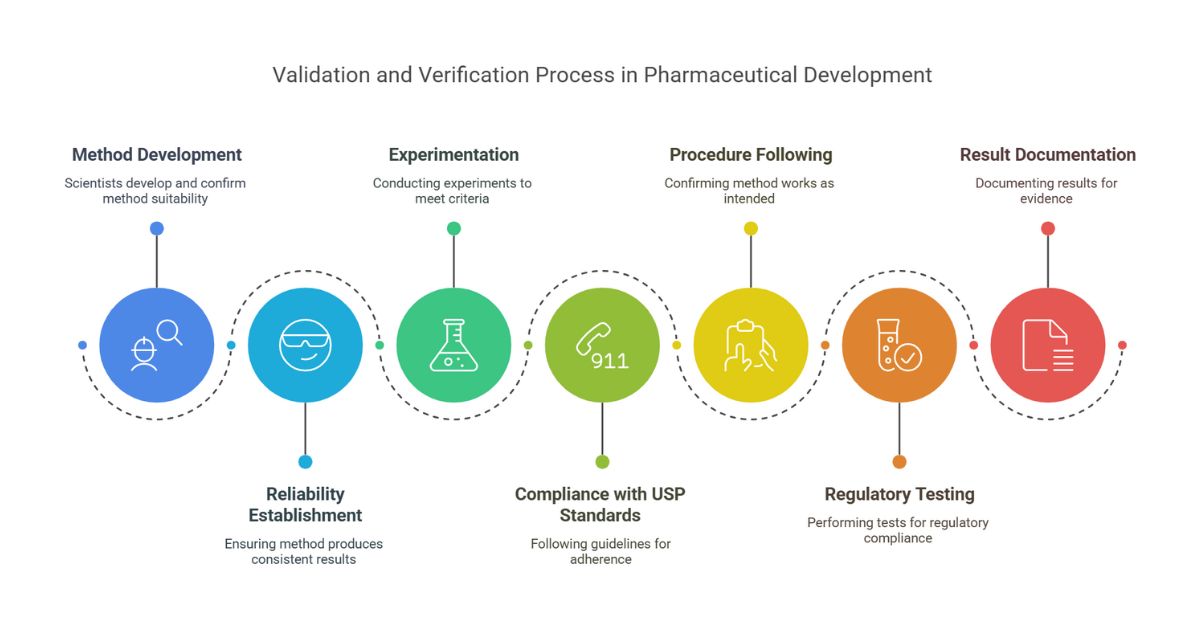
Method validation is the process of proving that an analytical method (e.g., testing drug purity, potency, or dissolution) is reliable, reproducible, and suitable for its intended use. Regulatory bodies like the FDA, ICH, and USP require it to ensure drugs are safe, effective, and consistent.
Why Pharma Labs Can’t Skip Validation:
Ensures consistency across batches and manufacturing sites.
Prevents recalls, lawsuits, and patient harm.
Meets strict FDA/ICH guidelines (e.g., ICH Q2(R2), USP <1225>).
Method Validation vs. Verification in Pharma
| Aspect | Method Validation | Method Verification |
|---|---|---|
| When Used | Developing a new method (e.g., for a novel drug). | Adopting a compendial method (e.g., USP monograph). |
| Regulatory Focus | ICH Q2(R2), FDA Guidance for Industry. | USP <1226>, FDA 21 CFR Part 211. |
| Complexity | Extensive (tests all validation parameters). | Tests lab-specific factors (equipment, analysts). |

The 7-Step Pharma Method Validation Process
Define the Method’s Purpose
- What are you testing? Examples:
- Potency: HPLC method for assay determination.
- Impurities: UPLC-MS/MS or GLC for detecting genotoxic impurities.
- Dissolution: UV-Vis method for tablet release.
- Set acceptance criteria based on ICH Q2(R2) and product specifications.
Risk Assessment
- Use ICH Q9 principles to identify critical parameters (e.g., column temperature variability in HPLC).
Design Experiments
- Test parameters like accuracy, precision, and robustness (see table below).
- Use pharmaceutical reference standards (e.g., USP-grade APIs).
Conduct Testing
- Test under “worst-case” scenarios:
- Different analysts, equipment, days.
- Deliberate variations (pH, flow rate, column age).
Analyze Data Statistically
- Tools: % recovery, confidence intervals, control charts.
- Example: A potency method passes if recovery is 98–102%.
Document in a Validation Report
- Include raw data, deviations, and corrective actions.
Review & Approve
- QA signs off before the method enters routine use.
Critical Validation Parameters for Pharma Methods
| Parameter | What It Ensures | Pharma Example |
|---|---|---|
| Accuracy | Results match true values (e.g., API concentration). | Spiking a placebo with 100mg API and recovering 98–102mg. |
| Precision | Consistency across repeats, days, analysts. | Relative Standard Deviation (RSD) <2% for assay. |
| Specificity | No interference from excipients, degradants, or matrix. | HPLC method distinguishes API from 5 degradants. |
| Linearity | Direct correlation between analyte and signal. | The method works despite small, intentional changes. |
| Robustness | Method works despite small, intentional changes. | Varying HPLC flow rate (±0.1 mL/min) doesn’t affect results. |
| LOQ/LOD | Quantifies/detects trace impurities. | LOQ = 0.05% for genotoxic impurities. |
Pharma-Specific Challenges & Fixes
Challenge 1: “If dissolution method fails during scale-up!”
Solution: Revalidate using biorelevant media and consider hydrodynamics in large vessels.
Challenge 2: “Impurity peaks overlap with the API in HPLC.”
Solution: Optimize chromatographic conditions (gradient, column type) or use mass spectrometry.
Challenge 3: “FDA flagged our validation during an audit.”
Solution: Align with ICH Q2(R2) 2023 updates and include forced degradation studies.
Best Practices for Pharma Labs
- Use Stability-Indicating Methods: Prove the method detects degradants (perform forced degradation under heat, light, and pH stress).
- Leverage Design of Experiments (DoE): Systematically test variables (e.g., DOE for HPLC method development).
- Control Environmental Factors: Temperature/humidity can alter hygroscopic drug results.
- Revalidate When:
- Changing manufacturers for excipients/APIs.
- Upgrading equipment (e.g., HPLC to UPLC).
- Moving production to a new facility (method transfer).
FAQs:
How much sample is needed for validation?
That will be enough for triplicate tests across all parameters. For impurities, include spiked samples.
Do clinical trial methods need full validation?
Phase 1 methods require partial validation (accuracy, precision). However, Phase 3 needs full validation.
What’s the biggest FDA audit red flag?
Missing robustness data. Regulators want proof your method works under stress conditions.
Can I validate a method for multiple products?
Only if they’re identical in formulation like tablets of the same composition or the same API/excipient of different vendors.
How to handle OOS (Out-of-Specifications) results during validation?
Investigate root causes (equipment? analyst error?) and document corrective actions. Other wise repeat the process.
References:
- ICH Topic Q 2 (R1) Validation of Analytical Procedures: Text and Methodology [European Medicines Agency-June 1995 CPMP/ICH/381/95 ]

")

