In pharmaceutical manufacturing, aseptic processing control is the distinct methodology used to produce sterile drug products that cannot be terminally sterilized due to thermal instability. Unlike terminal sterilization, which destroys microorganisms in the final sealed container, aseptic processing relies on excluding microorganisms from the process entirely. This requires a complex interplay of sterilized components, strictly controlled environments, and highly trained personnel.
As we move through 2025, the stakes for contamination control have never been higher. The global harmonization of GMP standards—particularly the emphasis on a holistic Contamination Control Strategy (CCS)—has shifted the industry from reactive monitoring to proactive risk prevention. A single lapse in aseptic technique can lead to product recalls, severe regulatory enforcement actions, and, most critically, patient harm.
The objective of this article is to provide a definitive, expert-level resource for awareness, compliance, and operational excellence. We will synthesize the latest expectations from the US FDA, WHO, Health Canada, and leading technical bodies to outline the must-have strategies for modern aseptic facilities.
Current Regulatory Landscape (2025)
The regulatory framework governing sterile manufacturing has evolved to demand “built-in” quality rather than reliance on end-product testing.
Key Regulatory Pillars
- US FDA (Guidance for Industry – Sterile Drug Products): The FDA emphasizes that sterility is assured not by testing, but by the validation of the manufacturing process. The agency requires robust design in facilities (e.g., separate areas for dynamic operations), validated sterilization cycles for all equipment/components, and rigorous personnel training.
- WHO Good Manufacturing Practices (Annex 6): Recent updates have harmonized significantly with global standards (like EU Annex 1), placing central importance on the CCS. It mandates specific grades for cleanrooms (Grades A, B, C, D) and details precise expectations for barrier technologies.
- Health Canada (GUI-0119): This guidance provides granular expectations for environmental and process monitoring, stressing that monitoring programs must be designed based on risk assessment to detect any drift from validated states.

The Shift to Contamination Control Strategy (CCS)
A major shift in 2025 is the explicit requirement for a formal CCS. As detailed in industry analyses, a CCS is not a single document but a “living” master plan that links all control measures—from raw material receipt to final product release. It must identify all critical control points (CCP) and assess the effectiveness of mitigations.
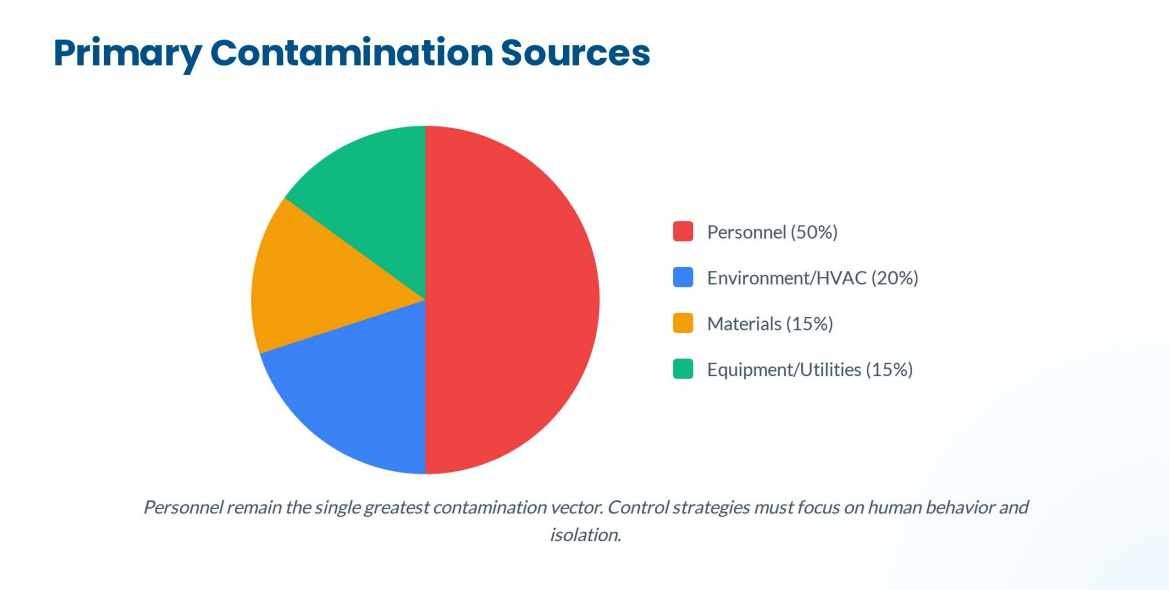
Sources and Types of Contamination
Understanding the enemy is the first step in prevention. Contamination in an aseptic area generally falls into three categories: viable (microorganisms), non-viable (particles), and chemical.
Primary Sources
Based on industry guidelines, the main vectors include:
- Personnel: The single greatest source of contamination. Humans shed thousands of skin cells and carry microorganisms despite rigorous gowning.
- Facility and HVAC Systems: Improper pressure cascades, inadequate High-Efficiency Particulate Air (HEPA) filtration, or poor airflow design can introduce and spread contaminants.
- Material Transfer: Every item entering Grade A/B areas is a potential carrier if not properly disinfected or sterilized.
- Equipment and Utensils: Improperly cleaned or sterilized product-contact surfaces.
- Process Utilities: Water for Injection (WFI) systems, steam, and compressed gases can breed biofilm or introduce particulates if not strictly managed.

Facility and Cleanroom Design for Aseptic Control
Modern facility design focuses on segregation and aerodynamic protection of the critical zone.
Zoning and Airflow
- Critical Area (Grade A/ISO 5): Must maintain strictly defined particle and microbial limits. FDA and ISO guidelines emphasize unidirectional airflow (laminar flow) at velocities sufficient to sweep particles away from the product (typically 0.36 – 0.54 m/s).
- Supporting Areas (Grade B/C/D): These act as successive buffers. Crucially, robust pressure differentials (typically at least 10-15 Pascals between differing classifications) must be maintained to prevent ingress of lower-quality air into higher-grade areas.
Barrier Technologies: RABS vs. Isolators
The 2025 standard increasingly favors advanced barrier technologies over classic cleanrooms to separate personnel from the process.
- Isolators: Offer the highest level of protection by completely sealing the process from the surrounding environment. They permit automated bio-decontamination (e.g., VHP) and generally allow for less rigorous background environments (e.g., Grade C or D, depending on design).
- Restricted Access Barrier Systems (RABS): Provide a rigid wall enclosure and utilise glove ports, but are not fully sealed (they rely on dynamic airflow for protection). RABS must be operated in a high-grade (Grade B) background.
- Selection: Isolators are preferred for high-potent drugs and maximum sterility assurance, while RABS offer flexibility and faster changeovers but require stricter surrounding operational controls.

Source: www.containedairsolutions.co.uk
Personnel & Aseptic Behavior Control
Because operators are the primary contamination risk, their management is paramount.
- Gowning: Must be sterile, non-shedding, and cover all skin virtually completely for Grade A/B entry. Gowning qualification is required before an operator can enter aseptic areas.
- Aseptic Technique:
- Slow, Deliberate Movement: Rapid motion creates turbulence that can overcome unidirectional airflow.
- “First Air” Principle: Never obstruct the path of sterile air between the HEPA filter and the critical product/component surface.
- Minimal Intervention: Modern processes are designed to minimize or eliminate inherent manual interventions.
Environmental and Process Monitoring
Monitoring does not assure sterility but verifies that the process is under control.
Monitoring Program Elements (GUI-0119 & WHO Annex 6)
- Viable Monitoring: Involves active air sampling (volumetric), passive air sampling (settle plates), surface contact plates, and personnel monitoring (gloves/garments).
- Non-Viable Particulate Monitoring: Continuous real-time monitoring is standard for Grade A areas during operations to detect immediate excursions.
- Trend Analysis: 2025 expectations move beyond just reacting to alert/action level excursions. Facilities must analyze data trends over time to detect slow drifts in facility performance before they become failures.
Aseptic Process Simulation (Media Fill) and Validation
A media fill is the ultimate validation of the aseptic process, substituting a sterile microbiological growth medium for the actual product.
- Frequency & Design: FDA and global standards typically require media fills semi-annually for each processing line and shift. The design must incorporate “worst-case” scenarios, including maximum allowed personnel, all permitted interventions, and longest run durations.
- Acceptance Criteria: The target is always zero contaminated units. Any positive unit indicates a potential failure in the sterility assurance system and requires a thorough investigation, often leading to a repeat of the stipulated number of successful runs (usually three) to requalify the line.
- PET Drugs Special Case: FDA guidance notes specific considerations for Positron Emission Tomography (PET) drugs due to their short half-lives, sometimes allowing for different validation approaches suitable for these rapid, small-batch processes.
Cleaning, Disinfection, and Sterilization
Effective contamination control relies on a validated cleaning and disinfection program.
- Distinctions:
- Cleaning: Removal of physical soil/residues.
- Disinfection: Reduction of microorganisms to a safe level.
- Sterilization: Complete destruction of all viable microorganisms (Sal 10^-6).
- Disinfectant Rotation: It is a regulatory expectation to rotate disinfectants regularly, including the periodic use of a sporicidal agent, to prevent the development of resistant resident flora.
- Validation: Disinfectant efficacy must be validated in-house against normal facility flora (environmental isolates), not just standard compendial challenge organisms.
Advanced Risk Assessment and Control Strategies
Building a robust CCS requires advanced risk management tools (e.g., HACCP, FMEA) applied specifically to sterility assurance.
The CCS “Living Document”
A compliant CCS represents a cyclical process of:
- Identify Risks: Map the entire process for potential ingress points.
- Assess Controls: Are current HEPA filters, gowning procedures, and sterilization cycles sufficient?
- Monitor Performance: Use data from environmental monitoring and media fills to verify control.
- Review and Improve: Periodic review of the CCS is mandatory to address emerging trends or changes in guidance.
Innovations and Future Directions
The industry is trending toward “absolute” protection.
- Automation and Robotics: Removing the human element entirely from the critical zone using robotic arms for interventions within isolators.
- Rapid Microbiological Methods (RMM): Moving from traditional multi-day incubation to faster, sometimes real-time, detection technologies to allow quicker responses to contamination events.
Frequently Asked Questions (FAQs)
Q: How often must operators be requalified for aseptic gowning? A: Typically annually, though many best-practice facilities requalify more frequently or immediately following a critical monitoring excursion.
Q: Can RABS replace Isolators entirely? A: While RABS improve upon traditional cleanrooms, Isolators generally offer a higher Sterility Assurance Level (SAL) and are preferred by regulators for new facility designs, especially for potent or highly sensitive products.
Q: What is the most common cause of media fill failure? A: Personnel interventions and poor aseptic technique (e.g., blocking first air) are historically the most frequent root causes.
Conclusion
Aseptic processing in 2025 demands a holistic, vigilant mindset. Compliance is no longer about passing a singular inspection but maintaining a continuous state of control through a living Contamination Control Strategy. By integrating advanced barrier technologies, rigorous behavioral training, and proactive data trending, manufacturers can ensure patient safety and meet the increasingly stringent global regulatory expectations.
References
- US FDA. Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice.[ https://www.fda.gov/media/71026/download]
- US FDA. Guidance for Industry: Media Fills for Validation of Aseptic Preparations for Positron Emission Tomography (PET) Drugs. [https://www.fda.gov/media/71119/download]
- World Health Organization (WHO). Annex 6: WHO good manufacturing practices for sterile pharmaceutical products. [https://www.who.int/publications-detail-redirect/WHO-TRS-1025-annex-6]
- Health Canada. Good manufacturing practices guide for drug products (GUI-0001) – Annex 1 to the Good manufacturing practices guide – Manufacture of sterile drugs (GUI-0119). [https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/good-manufacturing-practices/guidance-documents/gui-0119-manufacture-sterile-drugs/document.html]
- American Pharmaceutical Review. Establishing a Contamination Control Strategy for Aseptic Processing. [https://www.americanpharmaceuticalreview.com/Featured-Articles/571983-Establishing-a-Contamination-Control-Strategy-for-Aseptic-Processing/]
- PDA. In-process Microbial Control During Aseptic Processing. [https://www.pda.org/pda-letter-portal/in-process-microbial-control-during-aseptic-processing]
- Pharma Guideline. Different Sources of Contamination in an Aseptic Area. [https://www.pharmaguideline.com/2013/12/sources-of-contamination-in-aseptic-area.html]
- Pharma Guideline. Contamination Control Strategies for Manufacturing Area. [https://www.pharmaguideline.com/2020/12/contamination-control-strategies-for-manufacturing-area.html]
- PharmaGMP.in. Effective Strategies to Prevent Cross-Contamination in Sterile Product Manufacturing. [https://pharmagmp.in/effective-strategies-to-prevent-cross-contamination-in-sterile-product-manufacturing/]
- ISO/ICCCS. ISO Contamination Control Standards. [https://www.icccs.net/iso-standards]
- 14644.dk. Best Practices for managing cleanroom contamination. [https://14644.dk/best-practices-for-managing-cleanroom-contamination]
- GMP SOP. What is Environmental Monitoring in Pharmaceutical Industry. [https://gmpsop.com/gmp-articles/what-is-environmental-monitoring-in-pharmaceutical-industry]
- Esco Pharma. RABS vs Isolators: Understanding the Differences. [https://www.escopharma.com/articles/view/id/5989/rabs-vs-isolators-understanding-the-differences]
- Syntegon. RABS vs. Isolator – Choosing the right isolation technology. [https://www.syntegon.com/about-us/news-press/news/rabs-vs-isolator-choosing-the-right-isolation-technology]
- Ascendia CDMO. Aseptic Pharmaceutical Manufacturing: How It Works. [https://ascendiacdmo.com/aseptic-pharmaceutical-manufacturing-how-it-works/]
- Emma International. Aseptic Technique in Sterile Drug Manufacturing. [https://emmainternational.com/aseptic-technique-in-sterile-drug-manufacturing/]
- ECA Academy. FDA Expectations for Media Fills. [https://www.gmp-compliance.org/gmp-news/fda-expectations-for-media-fills]
- GXP Cellators. Media Fill: A Critical Validation Process In Aseptic Manufacturing. [https://gxpcellators.com/media-fill-in-aseptic-manufacturing/]
- Pharma Guideline. Aseptic Filling Process (Media Fill) Validation Protocol. [https://www.pharmaguideline.com/2016/09/media-fill-validation-protocol.html]
- Zamann Pharma. Contamination Control Strategy: A Guide to Creating an Effective Plan. [https://zamann-pharma.com/en/contamination-control-strategy-guide/]

: MOA, Approvals & India")

