
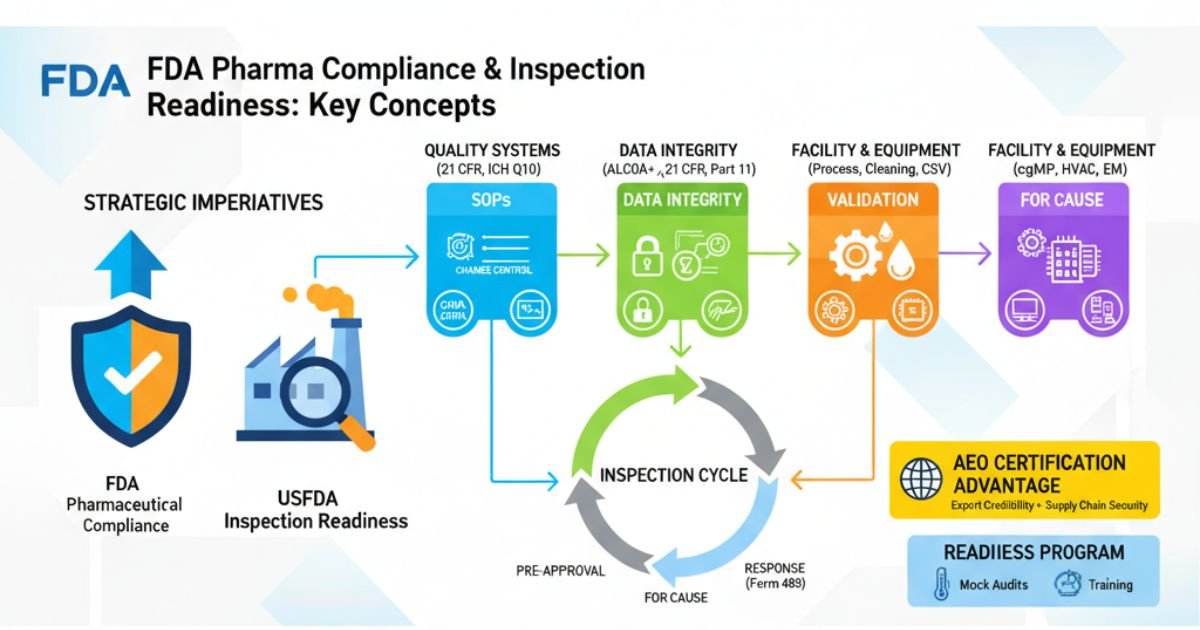
For pharmaceutical manufacturers, especially those eyeing the lucrative United States market, USFDA pharma compliance and Inspection readiness is not merely a regulatory hurdle; it is the cornerstone of commercial viability and corporate reputation. The U.S. Food and Drug Administration (FDA) acts as the gatekeeper to this market, and its enforcement of Current Good Manufacturing Practice (cGMP) regulations is rigorous, detailed, and increasingly global.
This guide serves as a comprehensive playbook for achieving and maintaining a state of USFDA inspection readiness. We will deconstruct the FDA’s regulatory framework, dissect common compliance failures, and provide actionable strategies to build a resilient Pharmaceutical Quality System (PQS). This includes a deep dive into data integrity (ALCOA+), process validation, risk management (ICH Q9), and responding to inspectional observations (Form 483).
Crucially, we will also explore the strategic advantage of Authorized Economic Operator (AEO) certification, a designation that enhances export credibility and signals a mature, secure supply chain—a factor of growing importance in the global regulatory landscape. The ultimate goal is to shift your organization’s mindset from reactive “preparation” to a proactive culture of “always ready,” where an unannounced FDA inspection is not a crisis, but an opportunity to demonstrate excellence.

The FDA Regulatory Framework: Understanding the Rules of the Road
Successfully navigating FDA expectations begins with a deep understanding of the regulations that form the basis of every inspection. These rules are not just a checklist; they represent a foundational philosophy for ensuring patient safety.
The Foundation of USFDA Compliance: 21 CFR Parts 210 & 211
The legal bedrock for pharmaceutical manufacturing in the U.S. is Title 21 of the Code of Federal Regulations (CFR). Specifically, Parts 210 and 211 outline the cGMP requirements:
- 21 CFR Part 210: Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or Holding of Drugs; General. This section lays out the general provisions and definitions.
- 21 CFR Part 211: Current Good Manufacturing Practice for Finished Pharmaceuticals. This is the core of the regulation, detailing the minimum requirements for everything from personnel and facilities to equipment, production controls, and laboratory testing.
Adherence to cGMP ensures that a drug product is safe, has the correct identity and strength, and meets the quality and purity standards it purports to have. Any drug manufactured in a facility that does not comply with cGMP is considered “adulterated” under U.S. law.
The Global Context: ICH Guidelines (Q7, Q9, Q10) and WHO-GMP
While 21 CFR is the U.S. law, the FDA’s thinking is heavily influenced by global harmonization efforts, primarily through the International Council for Harmonisation (ICH). Key ICH guidelines that every manufacturer should treat as essential reading include:
- ICH Q7 (Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients): The global standard for API manufacturing.
- ICH Q9 (Quality Risk Management): Provides a framework for making science-based, risk-based decisions about quality, a principle that is now central to the FDA’s inspectional approach.
- ICH Q10 (Pharmaceutical Quality System): Describes a comprehensive model for an effective PQS, integrating cGMP with concepts from ISO quality management systems.
Understanding these ICH guidelines is crucial, as they explain the “why” behind many of the FDA’s expectations for a modern, risk-based pharma quality system.
Four Types of FDA Inspections
An FDA inspection is not a monolithic event. The purpose of the visit dictates the investigator’s focus and scope. Understanding the type of inspection you are facing is the first step to managing it effectively.
Pre-Approval Inspection (PAI)
A PAI is conducted after a company submits a New Drug Application (NDA) or Abbreviated New Drug Application (ANDA) but before the product is approved for the market. The core question the FDA seeks to answer is: “Can this facility consistently manufacture this specific product according to cGMP and the application?”. The focus is intensely on manufacturing capability and, critically, the integrity of the data submitted in the application.
Surveillance (Routine GMP) Inspection
This is the most common type of inspection. It is a periodic, routine “check-in” to verify a facility’s ongoing compliance with cGMP regulations. The FDA uses a risk-based model to determine the frequency of these inspections, with higher-risk facilities (e.g., those producing sterile injectables) being inspected more often.
For-Cause Inspection
This inspection is triggered by a specific event or concern, such as a high volume of adverse event reports, a product recall, or a whistleblower complaint. These inspections are highly targeted, often unannounced, and focus on investigating the specific issue that prompted the visit.
Bioresearch Monitoring (BIMO) Inspection
The BIMO program focuses on the integrity of clinical and non-clinical research data. These inspections audit clinical trial sites, sponsors, and institutional review boards to ensure data submitted to the FDA is accurate and that human subjects were protected during trials.
The Core Pillars of cGMP Compliance
Achieving a state of USFDA inspection readiness requires more than just following procedures. It demands a demonstrable commitment to foundational quality principles, embodied in several interconnected pillars.
1: Pharmaceutical Quality Management System (PQS)
Your PQS is the formal system that designs, implements, monitors, and maintains all activities required to ensure product quality. It’s not just the Quality Assurance department; it’s an ecosystem encompassing all cGMP-related activities. A robust PQS, as described in ICH Q10, has:
- Strong Management Commitment: Leadership actively participates in and provides resources for the PQS.
- A Capable Quality Unit: The quality unit is independent, well-staffed, and has the authority to approve or reject materials and products.
- Clear Processes: All cGMP processes are clearly defined, documented, and controlled.
2: Documentation That Tells a Coherent Story
FDA investigators are trained to assess the health of your quality system through your documentation. The goal is to move beyond “archaeology-based documentation” where records are buried and disconnected. Instead, your documentation must tell a clear, logical, and complete story. An inspector should be able to pull any thread—a batch record, a deviation, a change control—and follow it logically through your system without needing verbal explanations or “tribal knowledge” to fill in the gaps.
3: Robust Validation (Process, Cleaning, Computer System)
Validation provides documented evidence that a process, method, or system consistently produces the expected result. It is a cornerstone of cGMP.
- Process Validation: Proves that your manufacturing process consistently yields a product of the required quality.
- Cleaning Validation: Demonstrates that cleaning procedures effectively remove residues to prevent cross-contamination.
- Computer System Validation (CSV): Ensures that software and computer-controlled systems perform accurately and reliably, with data integrity maintained.
4: Effective CAPA and Change Control Systems
No system is perfect. The FDA expects to see deviations, but what they scrutinize is how you manage them.
- CAPA (Corrective and Preventive Action): A strong CAPA system doesn’t just fix the immediate problem; it employs rigorous root cause analysis to understand why the failure occurred and implements changes to prevent it from happening again. Superficial investigations are a major red flag.
- Change Control: A formal system for managing any planned changes to facilities, equipment, processes, or documents. It ensures that changes are justified, documented, and assessed for their impact on product quality before implementation.
Data Integrity System
In the eyes of the FDA, the data your firm generates is a direct reflection of your control over your processes. Data integrity is the foundation upon which all regulatory trust is built; its failure is considered a fundamental breach of that trust.
Understanding ALCOA+ Principles
The governing standard for data integrity is the ALCOA+ framework. Data must be:
- Attributable: Who performed an action and when?
- Legible: Can you read the data throughout its lifecycle?
- Contemporaneous: Recorded at the time the work was performed.
- Original: The first recording of the data (or a true copy).
- Accurate: The data is correct and reflects what actually happened.
- + (Plus): The data must also be Complete, Consistent, Enduring, and Available.
The Critical Role of Audit Trails
For electronic systems, audit trails are non-negotiable. They provide a secure, time-stamped record of all actions related to creating, modifying, or deleting electronic data. However, simply activating an audit trail is not enough. A common FDA finding is the failure to routinely review audit trails. This review is critical for detecting unauthorized changes or potential data manipulation.
Data Integrity as a Cultural Imperative
Analysis of recent FDA Warning Letters to Indian firms reveals that “Lack of Data Documentation Discipline” and “Data Manipulation/Data Integrity” account for a combined 27% of all observations. This highlights that data integrity is not just an IT issue; it’s a cultural one. It requires training, clear procedures, and a management culture that has zero tolerance for poor documentation practices like backdating or sharing passwords.
Final Facility Readiness
Your physical facility is the container for your processes. Its design and state of maintenance are direct indicators of your commitment to cGMP.
Design, Layout, and Material/Personnel Flow
A well-designed facility minimizes the risk of mix-ups and contamination. Key principles include:
- Logical Flow: Materials and personnel should move through the facility in a logical, often unidirectional, flow to prevent cross-contamination.
- Adequate Space: There must be sufficient space for the orderly placement of equipment and materials.
- Segregation: Clear separation of areas for different functions, such as receiving, quarantine, manufacturing, and packaging. For sterile manufacturing, this includes strictly controlled classifications (Grade A, B, C, D) with validated pressure differentials.
HVAC, Environmental Monitoring, and Pest Control
- HVAC (Heating, Ventilation, and Air Conditioning): The HVAC system is critical for controlling air quality, temperature, humidity, and pressure differentials, especially in cleanrooms.
- Environmental Monitoring (EM): A robust, risk-based EM program is required to monitor for microbial and particulate contamination in controlled areas. The justification for sampling locations and frequencies must be scientifically sound. Inadequate smoke studies to visualize airflow patterns are a common citation in sterile facilities.
- Pest Control: A documented and effective pest control program is a basic cGMP requirement.
Process Validation and Continuous Verification
Process validation is the documented evidence that the manufacturing process, operated within established parameters, can effectively and reproducibly produce a medicinal product meeting its predetermined specifications and quality attributes.
The Validation Lifecycle: IQ, OQ, PQ
The traditional validation approach involves three stages:
- Installation Qualification (IQ): Verifying that the equipment has been installed correctly and according to the manufacturer’s recommendations and design specifications.
- Operational Qualification (OQ): Demonstrating that the equipment operates as intended throughout its specified operating ranges.
- Performance Qualification (PQ): Proving that the equipment, working together with personnel and procedures, consistently produces a quality product under normal operating conditions.
Process Performance Qualification (PPQ) and the Modern Approach
The FDA’s current thinking, outlined in its 2011 Process Validation Guidance, emphasizes a lifecycle approach. The key stage is Process Performance Qualification (PPQ), where the facility, utilities, equipment, and trained personnel are used to produce commercial-scale batches. The goal is to confirm the process design and demonstrate that the commercial manufacturing process performs as expected. This is typically followed by a program of Continued Process Verification to ensure the process remains in a state of control during routine production.
Cleaning Validation and Risk Management
Cleaning validation is essential to prevent cross-contamination between different products manufactured on the same equipment. The FDA expects a robust program that proves your cleaning procedures are effective and consistent.
Establishing Science-Based Acceptance Limits
You must define the maximum allowable carryover of residues (e.g., previous product API, cleaning agents) and establish acceptance criteria. These limits must be based on a scientific and toxicological assessment, not arbitrary numbers. Common approaches include using limits based on therapeutic dose, toxicity data (LD50), or a general limit (e.g., 10 ppm).
A Risk-Based Approach to Validation Strategy
A risk-based approach, aligned with ICH Q9, should be used. This involves:
- Grouping Strategy (“Matrixing”): Grouping similar products or equipment to reduce the total number of validation studies required. This must be scientifically justified.
- Identifying “Worst-Case” Scenarios: Validating the cleaning process for the most difficult-to-clean product or the hardest-to-clean equipment locations.
Quality Risk Management (QRM) – The ICH Q9 Guidelines
Modern FDA pharmaceutical compliance is driven by the principles of Quality Risk Management (QRM), as detailed in ICH Q9. This is the practice of using science- and risk-based approaches to make decisions about quality throughout the product lifecycle. Instead of treating all issues with equal weight, QRM allows a company to focus its resources on the areas that pose the greatest risk to patient safety and product quality.
Frequent FDA findings point to an incomplete application of QRM principles, such as the lack of a well-documented scientific rationale for design choices, process controls, or validation approaches.
Vendor Qualification and Raw Material Control
Your finished product is only as good as the materials you start with. A robust supplier qualification program is a cGMP requirement. This involves:
- Vendor Audits: Auditing critical suppliers of raw materials and components to ensure they meet quality standards.
- Quality Agreements: Establishing clear, written agreements that define the quality and regulatory responsibilities of both parties.
- Incoming Material Testing: Performing identity testing and other appropriate tests on all incoming raw materials to verify their quality and suitability.
Computer System Validation (CSV) and 21 CFR Part 11 Compliance
In the modern pharmaceutical plant, computer systems are ubiquitous. Computer System Validation (CSV) is the process of demonstrating that these systems work as intended in your specific environment. It is a critical component of ensuring data integrity.
All electronic records and electronic signatures must comply with 21 CFR Part 11. This regulation sets the requirements for:
- System Security: Limiting access to authorized individuals.
- Audit Trails: Secure, computer-generated, time-stamped audit trails that independently record user actions.
- Electronic Signatures: Ensuring electronic signatures are as legally binding as handwritten ones.
- Record Integrity: Protecting records from alteration or deletion.
Responding to a USFDA Form 483
At the conclusion of an inspection, if the investigator observed conditions that may violate cGMP, they will issue a Form FDA 483, “Inspectional Observations”. A 483 is not a final determination of non-compliance, but a list of concerns.
How you respond is critically important. You have 15 business days to submit a formal, written response. A weak or incomplete response can be more damaging than the observations themselves.
A strategic response, sometimes called a “Four Horizons” approach, should detail:
- Immediate Correction/Containment: What you did immediately to address the specific observation.
- Root Cause Investigation: A thorough investigation into why the failure happened.
- Systemic Evaluation: An assessment of where else this problem might exist in your facility.
- Preventive Enhancements: The systemic changes you will make to prevent recurrence across the entire organization.
Decoding FDA Warning Letter Trends: Learning from Common Failures
If the FDA finds the 483 response inadequate or the violations severe, it can escalate to an Official Action Indicated (OAI) classification and issue a Warning Letter. Analyzing these letters provides invaluable insight into the FDA’s current focus areas.
An analysis of Warning Letters issued to Indian firms between CY2022 and May 2025 revealed a consistent pattern of systemic failures in foundational quality systems.
| Nature of Observation | Percentage Share |
| Failure to Maintain Quality and Purity | 24% |
| Lack of Data Documentation Discipline | 21% |
| Lack of Hygiene | 21% |
| Compliance* | 15% |
| Inadequate investigation of critical deviations | 9% |
| Data Manipulation/Data Integrity | 6% |
This data clearly shows that the most severe enforcement actions stem from failures in basic cGMP execution—quality systems, documentation, and hygiene—rather than highly technical issues.
cGMP FDA Audit Checklist for Manufacturers
This checklist provides a high-level overview of key areas to assess during an internal or mock audit. It is not exhaustive but covers the primary domains of an FDA inspection.
| Category | Checklist Item | Status (Compliant / Needs Improvement) |
| Quality Management System | Is the Quality Unit independent and adequately resourced? | |
| Is there a formal process for management review of the PQS? | ||
| Are roles, responsibilities, and authorities clearly defined? | ||
| Personnel | Are all employees qualified through education, training, and experience? | |
| Is training effectiveness periodically assessed? | ||
| Are personnel health and hygiene procedures adequate? | ||
| Facility & Equipment | Is the facility design adequate to prevent mix-ups and contamination? | |
| Is equipment qualified (IQ/OQ/PQ) and calibrated on schedule? | ||
| Are cleaning procedures validated and followed? | ||
| Is the environmental monitoring program risk-based and effective? | ||
| Production & Process Controls | Are Master Batch Records (MBRs) detailed and approved? | |
| Are all critical process parameters identified and controlled? | ||
| Is the manufacturing process validated (PPQ)? | ||
| Are in-process controls established and followed? | ||
| Laboratory Controls | Are lab methods validated or verified? | |
| Are Out-of-Specification (OOS) results thoroughly investigated? | ||
| Are lab instruments calibrated and maintained? | ||
| Is there a formal stability testing program? | ||
| Documentation & Data Integrity | Are records contemporaneous, legible, and accurate (ALCOA+)? | |
| Are batch records complete and reviewed by the Quality Unit? | ||
| Are audit trails enabled and reviewed on electronic systems (21 CFR Part 11)? | ||
| Is there a secure system for record retention and archiving? | ||
| Materials Management | Is there a robust supplier qualification program? | |
| Are incoming materials quarantined and tested before use? | ||
| Are materials stored under appropriate conditions? | ||
| System Management | Is the CAPA system effective at identifying root cause and preventing recurrence? | |
| Is there a formal Change Control system? | ||
| Are complaints and deviations investigated and documented thoroughly? |
The AEO Advantage
For export-focused pharmaceutical companies, cGMP compliance is only part of the puzzle. Securing the global supply chain is equally critical. This is where Authorized Economic Operator (AEO) certification provides a powerful strategic advantage.
What is AEO Certification?
AEO is a globally recognized quality mark under the World Customs Organization (WCO) SAFE Framework of Standards. It certifies that a company’s role in the international supply chain is secure and that its customs controls and procedures are efficient and compliant. While cGMP focuses on the quality of the product, AEO focuses on the security and integrity of the supply chain used to transport it.
How AEO Complements cGMP Compliance for Pharma Exporters
Achieving AEO status signals to regulators like the FDA and customs authorities that your organization has a mature, robust, and well-documented system of controls that extends beyond the factory floor. The benefits include:
- Enhanced Credibility: AEO certification acts as an external validation of your company’s commitment to control and security, which can positively influence a regulator’s perception of your overall compliance culture.
- Supply Chain Security: The rigorous security criteria for AEO help protect against theft, diversion, and counterfeiting of pharmaceutical products—a major global health concern.
- Faster Customs Clearance: AEO status often leads to fewer physical inspections and faster clearance at international borders, ensuring that time-sensitive medicines reach patients without delay.
- Demonstrated Control: The documentation and process control required for AEO are highly complementary to cGMP requirements. A company that can master AEO standards is likely to have the systemic discipline needed for robust cGMP compliance.
For a pharma exporter, holding both cGMP compliance and AEO certification presents a powerful narrative: “We manufacture a high-quality product under strict cGMP controls, and we ship it via a secure, validated AEO supply chain.”
Strategic framework for Building Inspection Readiness Program:
True USFDA inspection readiness is not a project; it is a continuous state of being. This is achieved through a structured, proactive program.
The Role of Mock Audits and Internal Audits
The most effective way to test your readiness is to simulate the real thing.
- Internal Audits: A routine program of self-inspection to identify and correct gaps before an external auditor finds them.
- Mock FDA Audits: These should be rigorous, unannounced audits conducted by experienced auditors (internal or external) who can accurately mimic the behavior and focus of an FDA investigator. The goal is to test your systems, document retrieval, and SME interview performance under realistic pressure.
Establishing a Cross-Functional Readiness Team
Create a dedicated, cross-functional inspection readiness team with representatives from Quality, Operations, Engineering, Regulatory Affairs, and R&D. This team is responsible for:
- Managing the Program: Overseeing the internal and mock audit schedules.
- SOP Development: Creating and maintaining robust SOPs for managing an inspection (e.g., investigator reception, document control “war room,” SME escorting).
- Training: Ensuring all personnel, especially SMEs, are trained on how to interact with investigators.
Best Practices for Compliant:
- Train for the “Why”: Shift training from rote procedural memorization to a deep understanding of the scientific and regulatory rationale behind an action. An operator who can explain why a parameter is critical is far more convincing than one who just follows an SOP.
- Practice “Constructive Transparency”: No facility is perfect. When an issue is found, don’t hide it. Acknowledge it clearly and focus the discussion on your robust investigation, corrective actions, and effectiveness checks. A well-managed problem is a sign of a healthy quality system.
- Embed a “Why-First” Investigation Framework: Mandate the use of robust root cause analysis tools that compel teams to move beyond superficial conclusions and identify true systemic vulnerabilities.
- Shift from Lagging to Leading Indicators: Don’t just track failures (deviation counts). Proactively monitor leading indicators like process capability trends (Cpk), near-miss reports, and right-first-time rates to identify and mitigate risks before they become deviations.
90-Day FDA Compliance Improvement Roadmap
This is an aggressive but achievable plan for significantly enhancing your site’s inspection readiness in three months.
Phase 1: Diagnosis & Planning (Days 1-30)
- Assemble the Team: Form your cross-functional inspection readiness team.
- Conduct a Deep-Dive Gap Analysis: Perform a comprehensive internal audit against 21 CFR 211 and the checklist in this guide. Be brutally honest.
- Prioritize Findings: Use risk management (ICH Q9) to rank all identified gaps based on their potential impact on product quality and patient safety.
- Develop a Master CAPA Plan: Create a detailed project plan with specific actions, responsible owners, and firm deadlines for addressing every prioritized gap.
Phase 2: Remediation & Implementation (Days 31-60)
- Execute the CAPA Plan: Systematically close out the actions identified in Phase 1. This includes updating SOPs, retraining personnel, and implementing new controls.
- Focus on Data Integrity: Conduct a specific audit of your data governance practices (both paper and electronic). Review audit trails and system access controls.
- Strengthen “The Story”: Review key documentation systems (e.g., batch records, deviations) to ensure they are clear, logical, and interconnected.
- Conduct SME “Bootcamps”: Begin training key personnel on how to confidently and accurately answer questions from an investigator.
Phase 3: Verification & Sustenance (Days 61-90)
- Conduct a Mock FDA Audit: Hire an external consultant or use a qualified internal team to conduct a full, unannounced mock inspection to test your implemented changes under pressure.
- Refine Based on Mock Audit: Address any findings from the mock audit with the same rigor as a real one.
- Finalize Inspection Logistics: Ensure your inspection management SOPs are finalized and all personnel are trained on their roles (e.g., scribes, runners, backroom leads).
- Launch a Continuous Readiness Program: Transition from the 90-day project to a continuous program of daily walkthroughs, regular self-audits, and ongoing training to maintain the state of readiness.
Top 10 Common Mistakes that Attract FDA Scrutiny
- Superficial Investigations: Blaming “human error” without digging for the true systemic root cause.
- Poor Documentation Practices: Missing signatures, backdating, using white-out, or incomplete records.
- Inadequate Training: Personnel cannot explain the “why” behind their actions.
- Ignoring Data Integrity: Failing to review audit trails or having weak password controls.
- Unvalidated Processes: Lack of validation for cleaning, manufacturing processes, or computer systems.
- Poor Change Management: Implementing changes without a formal review and approval process.
- Deficient Laboratory Controls: Not investigating OOS results properly or using uncalibrated instruments.
- Disorganized Facility: A cluttered, dirty, or poorly maintained facility signals a lack of control.
- Ineffective CAPA System: “Closing” CAPAs without verifying that the actions were actually effective in preventing recurrence.
- A Disconnected “Story”: When batch records, deviations, and CAPAs don’t logically link together, forcing reliance on verbal explanations.
Frequently Asked Questions
What is the difference between cGMP and GMP?
“cGMP” stands for Current Good Manufacturing Practice. The “c” emphasizes that the standards are dynamic and that companies must use up-to-date technologies and systems to comply with the regulations.
How often does the FDA inspect pharmaceutical facilities?
The frequency is risk-based. High-risk facilities, like those making sterile products, may be inspected every two years, while lower-risk facilities may have a longer interval. For-cause inspections can happen at any time.
Can the FDA conduct unannounced inspections outside the U.S.?
Yes. In recent years, the FDA has explicitly stated its intent to increase the use of unannounced inspections for foreign facilities to ensure a state of “real-time compliance”.
What is the most common citation in FDA 483s?
Consistently, one of the most cited regulations is 21 CFR 211.22(d), related to the Quality Control unit’s responsibility for investigating deviations and failures. This points to widespread issues with investigations and CAPA systems.
What is ALCOA+ and why is it important for FDA data integrity?
ALCOA+ is an acronym (Attributable, Legible, Contemporaneous, Original, Accurate + Complete, Consistent, Enduring, Available) that defines the key characteristics of reliable data. It is the framework the FDA uses to assess data integrity.
Do I need to validate off-the-shelf software used in my lab?
Yes. All computer systems used in a GxP environment must be validated for their intended use. This is known as Computer System Validation (CSV).
How long do I have to respond to an FDA Form 483?
You have 15 U.S. business days to provide a formal written response to the FDA.
What is the difference between a Form 483 and a Warning Letter?
A Form 483 is a list of inspectional observations noted by the investigator on-site. A Warning Letter is a formal notification from senior FDA leadership indicating serious violations that require prompt and comprehensive corrective action.
How can a mock FDA audit help my company?
A mock audit tests your systems, procedures, and personnel in a realistic but low-stakes environment. It identifies gaps, builds staff confidence, and is the single best way to prepare for a real inspection.
What is Quality Risk Management (QRM) in pharma?
QRM (ICH Q9) is a systematic process for the assessment, control, communication, and review of risks to the quality of the drug product. It helps companies focus resources on the highest-risk areas.
What is a Contamination Control Strategy (CCS)?
A CCS is a holistic, risk-based strategy to control contamination in sterile manufacturing facilities. It integrates facility design, personnel training, process controls, and environmental monitoring into a single, cohesive system.
What is the FDA PreCheck program?
Announced in 2024, the FDA PreCheck program is a U.S. initiative designed to encourage domestic manufacturing by providing a pre-certification of a facility’s compliance, potentially streamlining approvals and oversight.
Why does the FDA focus so heavily on sterile manufacturing?
Sterile products are typically injected directly into the body, bypassing its natural defense mechanisms. Any contamination can lead to severe patient harm or death, so these products face the most stringent regulatory scrutiny.
Are small pharma companies held to the same cGMP standards as large ones?
Yes. The cGMP regulations apply to all manufacturers regardless of size. However, data suggests small to mid-sized firms may face resource constraints that make compliance more challenging, as a majority of recent Warning Letters were issued to them.
What is AEO certification and is it relevant for FDA compliance?
AEO (Authorized Economic Operator) certifies the security of a company’s international supply chain. While not an FDA requirement, it complements cGMP by demonstrating a culture of control and security that enhances regulatory credibility for pharma exporters.
Conclusion:
The landscape of FDA pharmaceutical compliance is evolving. With the rise of unannounced foreign inspections and increasingly complex global supply chains, the old model of “cramming for the audit” is obsolete. The fundamental question for today’s quality leaders is no longer ‘Are we compliant today?’ but rather, ‘Is our quality culture sufficiently robust to treat an unannounced FDA inspection as a routine event?’.
Achieving this state of USFDA inspection readiness requires a cultural transformation. It means embedding quality into daily operations, empowering personnel with a deep understanding of the “why,” and building systems that tell a coherent, data-driven story of control. By embracing the principles and strategies outlined in this guide—from mastering ALCOA+ and QRM to leveraging the credibility of AEO—manufacturers can move beyond mere compliance. They can build a resilient organization that protects patients, satisfies regulators, and secures a lasting competitive advantage in the global market.
Contact Us for a Comprehensive Compliance Audit
Navigating the complexities of FDA regulations can be daunting. Our team of former FDA investigators and senior industry consultants can help you build a culture of proactive compliance.
Contact us today for:
- Comprehensive Mock FDA Audits
- 483 & Warning Letter Response Strategy
- Data Integrity Gap Assessments & Remediation
- AEO Certification Consulting for Pharma Exporters
Let us help you turn your next FDA inspection into a showcase of your commitment to quality.
References
- Code of Federal Regulations (CFR) Title 21, Part 210 – Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or Holding of Drugs; General. [
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-210] - Code of Federal Regulations (CFR) Title 21, Part 211 – Current Good Manufacturing Practice for Finished Pharmaceuticals.[
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211] - FDA Guidance for Industry: Process Validation: General Principles and Practices. [
https://www.fda.gov/media/71021/download] - FDA Guidance for Industry: Data Integrity and Compliance With Drug CGMP: Questions and Answers. [
https://www.fda.gov/media/119229/download] - International Council for Harmonisation (ICH) Q9 – Quality Risk Management. [
https://database.ich.org/sites/default/files/Q9_Guideline.pdf] - International Council for Harmonisation (ICH) Q10 – Pharmaceutical Quality System.[
https://database.ich.org/sites/default/files/Q10_Guideline.pdf] - FDA MAPP 5010.1: Conduct of Pre-Approval Inspections. [
https://www.fda.gov/media/71036/download] - World Customs Organization (WCO) SAFE Framework of Standards to Secure and Facilitate Global Trade. [
https://www.wcoomd.org/-/media/wco/public/global/pdf/about-us/wco-safestandards-framework-to-secure-and-facilitate-global-trade_en.pdf]


: A Potential Breakthrough in Diabetes Treatment")
