
For global pharmaceutical and medical device manufacturers, particularly those operating in high-volume markets like the US and India, the receipt of an FDA Form 483, Inspectional Observations, is not merely a documentation step—it is a critical inflection point. In the intensified, risk-based regulatory climate of 2025, a weak, defensive, or untimely 483 response is almost a direct invitation for an escalating enforcement action, such as a Warning Letter, Import Alert, or even a Consent Decree.
The USFDA’s expectation has profoundly shifted. It is no longer enough to simply correct the specific observation; the agency expects you to demonstrate an “anti-fragile” quality organization—one that emerges stronger and more resilient from an inspectional finding. Your written response, drafted and committed to by your senior leadership, serves as the “first impression” and a “visual demonstration of your site’s quality culture.”
This authoritative guide synthesizes the deepest insights from FDA policy, enforcement trends, and industry best practices to provide a definitive, actionable, 7-Phase strategy that transcends mere compliance and establishes a robust, sustainable quality posture.
The New FDA Compliance Landscape
To write a compelling response, you must first understand the inspector’s current focus. The FDA’s strategy in 2025 is characterized by precision, data-driven targeting, and an unforgiving scrutiny of systemic failures.
Targeted Enforcement and AI-Driven Inspection
Routine inspections are being replaced by highly targeted inspections identified by sophisticated tools, such as the agency’s internal AI/machine learning analytics (often analyzing complaint trends, import data, and historical inspection outcomes). This means when an inspector arrives, they are usually already focused on a known, high-risk vulnerability in your system.
CAPA and Data Integrity (DI)
Data from recent fiscal years confirm the persistent criticality of these two areas:
| Top-Cited GMP Violation | Core Regulatory Failure |
| Corrective and Preventive Action (CAPA) | Insufficient root cause analysis, lack of effectiveness checks (verification), and failure to extend the CAPA scope enterprise-wide. |
| Data Integrity (DI) | Failures to meet ALCOA++ principles (Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, Available). Lack of secure audit trails, uncontrolled access, and “quick fixes” that don’t address systemic computerized system validation. |
| Aseptic Processing/Facility Controls | Repeat findings often linked to failed investigations of environmental monitoring (EM) excursions or cross-contamination risks, especially in multi-product facilities. |
Accountability for Third-Parties (CMOs)
The FDA is enforcing heightened accountability for sponsors concerning the quality systems of their Contract Manufacturing Organizations (CMOs). A failure at your CMO is a failure of your Purchasing Controls (21 CFR 211.84/820.50). Your 483 response must demonstrate robust supplier qualification, monitoring, and corrective action oversight for any observations cited at a third-party site.
USFDA Form 483 Response Strategy Workflow: A 7-Phase Strategy
A compliant, compelling 483 response must be executed with surgical precision and governed by strict timelines. This 7-Phase workflow ensures all FDA expectations—systemic CAPA, root cause, and product impact—are met.
Phase 1: Immediate Containment & Team Formation (Day 0–3)
The clock starts the moment the 483 is issued. The goal is to acknowledge the observations, contain the immediate risk, and establish a high-level command structure.
- Exit Meeting Deep Dive: During the close-out meeting, listen actively. Seek polite clarification, not confrontation. Ensure you fully understand the investigator’s interpretation and the specific regulatory clause cited. If a correction can be made during the inspection (e.g., immediate cleanup, procedural correction), implement it immediately and provide objective evidence before the inspector leaves.
- Establish the War Room: Form a multi-disciplinary Response Team (RT) led by a senior executive (e.g., Head of QA/VP of Operations) to ensure top-down commitment. The RT must include QA/QC, Operations, Regulatory Affairs, Engineering, and Legal Counsel (for complex/Warning Letter-risk observations).
- Initial Triage and Risk Tiers: Categorize the observations based on potential patient risk and regulatory impact (e.g., Critical – Data Integrity, Sterility Assurance; Major – CAPA failures, Out-of-Specification Investigations; Minor – Documentation errors). This dictates resource allocation and CAPA prioritization.
Phase 2: The Deep-Dive Root Cause Analysis (Day 3–7)
The failure of most 483 responses stems from a superficial root cause analysis (RCA). The FDA demands that you move beyond the symptom (the observation) to the systemic failure (the root cause).
- Systemic Investigation Scope: For every observation, the investigation must be enterprise-wide. Ask: Does this system failure affect other products, processes, or sites? (The “Broader Impact Assessment”).
- Formal RCA Methodology: Do not rely on informal methods. Use formalized tools, documented in a dedicated investigation report:
- 5 Whys: Excellent for simple, human-error observations.
- Fishbone (Ishikawa) Diagram: Essential for complex process/system failures (Man, Method, Machine, Material, Measurement, Mother Nature).
- Failure Mode and Effects Analysis (FMEA): Best practice for high-risk, systemic issues (e.g., aseptic processing) to prioritize future preventive actions.
- The “Human Element” Gap: A common mistake is citing “Human Error” as the root cause. This is never sufficient. The true root cause is the systemic failure that allowed the human error (e.g., inadequate training, poor procedure design, lack of visual cues, ineffective supervision).
Phase 3: Product Impact Assessment (Day 7–10)
This is a frequently understated requirement. You must thoroughly determine the impact of the observed non-conformance on the Safety, Identity, Strength, Purity, and Quality (SISPQ) of the affected product(s).
- Retrospective Review: Review all product lots potentially impacted by the systemic failure, extending back to the last compliant event or validation.
- Disposition & Control: Clearly state the disposition of all affected inventory (e.g., Product Hold on existing stock, Quarantine of intermediates, Recall Assessment). If the assessment is ongoing, provide an explicit, short-term commitment date for completion. Never delay this assessment.
Phase 4: Crafting the Systemic CAPA (Day 10–12)
The Corrective Action Plan must be specific, measurable, achievable, relevant, and time-bound (SMART).
- Immediate Correction (Correction): The fix to address the specific example cited in the 483. (e.g., “The SOP was revised and issued on 10/25/2025.”)
- Corrective Action (CA): The action addressing the root cause to prevent recurrence at the local level. (e.g., “All QA personnel received retraining on the revised SOP (documented training logs attached), and the electronic system access controls were updated (Change Control #CC-2025-045 initiated).”)
- Preventive Action (PA): The action addressing the systemic root cause to prevent the problem across the entire QMS/organization. (e.g., “A gap analysis was conducted across all other site SOPs and three similar control deficiencies were identified. A site-wide Corrective Action was initiated to revise those procedures by 12/15/2025.”)
Phase 5: Drafting & Review (Day 12–14)
The response must adhere to the 5 Cs of a Compelling Response:
| Characteristic | Requirement | Impact |
| Clear | Use plain, unambiguous language. Restate the observation exactly as written. | Avoids misinterpretation; shows acknowledgment. |
| Concise | Avoid unnecessary details, overly verbose explanations, or “defensive” text. | Respects the reviewer’s time; gets directly to the solution. |
| Compelling | Provide objective evidence (proof of completed actions) and demonstrate a firm commitment to future actions. | Establishes credibility; shows the problem is already being solved. |
| Complete | Address every part of every observation, including the systemic impact. Commit to all necessary CAPAs, timelines, and ownership. | Prevents follow-up questions; reduces risk of Warning Letter escalation. |
| Committed | Ensure the cover letter is signed by a senior executive with corporate-level responsibility. | Demonstrates top-down accountability and resources dedication. |
Phase 6: Submission & Timeline Discipline (Day 15)
The 15-business-day rule is not a legal mandate, but it is a definitive policy expectation. Do not miss it.
- Initial Response (Mandatory): Even if all CAPAs are not complete, submit a comprehensive, committed response within 15 days, providing the detailed plan and completion dates for ongoing actions.
- Follow-Up Responses: If a CAPA has a long implementation timeline (e.g., facility renovation), explicitly commit to providing periodic updates (e.g., “A status update will be provided every 30 days until the CAPA is closed.”). This proactive communication shows control and commitment.
Phase 7: Effectiveness Monitoring (Ongoing)
The lifecycle of a CAPA is not complete until its effectiveness is verified.
- Effectiveness Checks: For every CAPA, define how you will measure its success (e.g., no recurrence of the observation in internal audits for 12 months; trending of related quality events drops by 50%).
- Internal Audit Integration: Integrate the post-483 remediation plan into the next cycle of internal and vendor audits. A repeat finding is a near-guarantee of a Warning Letter, as it demonstrates a fundamental failure of your corrective action system.
")
Advanced Guidance: Handling Complex Observations
Certain observations carry disproportionately high regulatory risk and require a uniquely specialized response.
1. Data Integrity (DI) Observation Response
DI citations (such as failure of audit trails, shared passwords, or unreviewed critical data) are often considered tantamount to fraud and are handled by the agency’s sophisticated Office of Manufacturing Quality (OMQ).
- Go Beyond Technical Fixes: Do not just fix the software. The response must address the cultural element. Did personnel feel pressure to produce “right” results?
- ALCOA++ Implementation: Your CAPA must explicitly reference how systems and procedures now satisfy the extended ALCOA principles: Available, Consistent, Complete, and Enduring data. This includes secure, validated audit trails that are routinely reviewed as part of the QC unit’s responsibilities.
- Retrospective Data Review: Commit to a rigorous, documented review of all data generated by the affected system(s) to determine the extent of the DI compromise and the potential impact on product release decisions.
2. Repeat Findings (The Cardinal Sin)
A repeat 483 observation (a violation cited on a previous 483 or Warning Letter) signals a fundamental breakdown in your QMS—specifically, the failure of the prior CAPA.
- Response Strategy: Do not argue. Acknowledge the failure of the previous corrective action and propose a total system overhaul.
- Systemic CAPA on CAPA: The new RCA must focus on why the first CAPA failed. Was the original root cause incorrect? Was the effectiveness check inadequate? Did management fail to provide resources? Your new CAPA is now a CAPA on the CAPA system itself.
3. Global Compliance Synergy (US FDA vs. EU/WHO GMP)
For multi-national firms (especially those in India serving both US and European markets), leverage regulatory differences to your advantage, while ensuring US expectations are met.
| Compliance Area | US FDA Approach (Focus) | EU/WHO GMP Approach (Focus) |
| Validation | Flexible, Lifecycle Approach (Process Design, Qualification, Continuous Verification). | Prescriptive, Structured DQ/IQ/OQ/PQ model. |
| Batch Release | Quality Unit responsible for product disposition. | Required final release by a Qualified Person (QP). |
| 483 Response | Evidence-Driven, Systemic response focused on CAPA effectiveness and senior management commitment. | Response often integrates directly with QMS improvement plans; regulatory response is less public facing than the US system. |
The Integration Strategy: When responding to an FDA 483, adopt the EU/WHO GMP rigor (e.g., the structured DQ/IQ/OQ/PQ mindset) to document and validate your fixes, which satisfies the FDA’s demand for comprehensive objective evidence. Use a global quality system approach (e.g., ICH Q10) to ensure fixes applied for the FDA are simultaneously rolled out to satisfy international requirements.
4. Managing CMO/Third-Party Issues
If the observation is related to a third-party (CMO, contract lab), your response cannot simply blame them.
- Acknowledge Sponsor Responsibility: State that you acknowledge full responsibility for the quality of the product, regardless of where the non-conformance occurred.
- Purchasing Control CAPA: Your CAPA must detail how your purchasing controls system failed to prevent or detect the CMO’s non-compliance. This includes:
- Revised audit frequency and scope.
- Enhanced incoming quality checks (IQC) for the material/service.
- A commitment to issue a formal Quality Agreement Addendum specifying new expectations and monitoring metrics.
5. Escalation: When to Engage Expert and Legal Counsel
If observations involve Data Integrity, potential criminal misconduct, severe product quality/safety issues requiring a full recall, or are repeat findings escalating toward a Consent Decree, legal and regulatory counsel must be engaged immediately to shape the response strategy and privileged investigations. Counsel ensures that the response meets both regulatory expectations and legal protection standards.
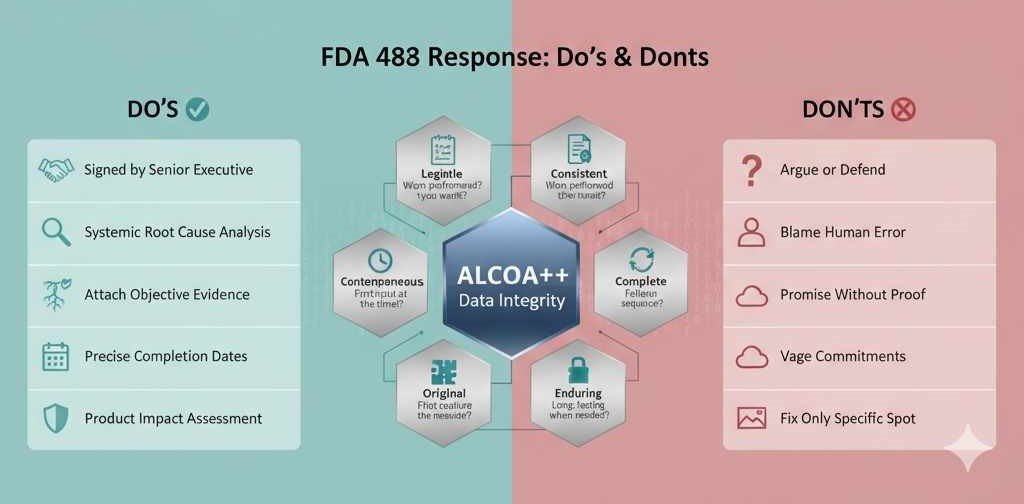
Actionable Do’s and Don’ts
Learning from past enforcement actions is the fastest route to compliance excellence.
| DO’S (The Successful Response) | DON’TS (The Failed Response) |
| DO restate the observation and provide a concise statement of agreement or factual clarification. | DON’T argue the semantics or defend the non-compliant process. It’s counterproductive. |
| DO include a signed cover letter from an officer with executive responsibility, committing corporate resources. | DON’T have a lower-level QA manager or consultant sign the cover letter—it signals a lack of senior commitment. |
| DO attach objective evidence (revised SOPs, training logs, validation protocols) for every completed action. | DON’T promise to train personnel without attaching the new training material and a summary of completed training records. |
| DO conduct a systemic/enterprise-wide RCA and CAPA, looking beyond the immediate issue/site. | DON’T cite “Human Error” as the root cause without addressing the system that enabled the error. |
| DO provide detailed, precise, and realistic completion dates for every CAPA element, clearly naming the owner. | DON’T use vague commitments like “will be addressed soon,” “improve compliance,” or “by Q1 next year” without a specific day/month. |
| DO assess and document the impact on all potentially affected product lots (SISPQ). | DON’T omit a formal product impact assessment or fail to place affected product on quality hold. |
")
Improved Structure & Checklist Response Template
An effective response is not a letter; it is a meticulously organized package.
I. Cover Letter (Executive Summary)
- Header: Dated, addressed to the Investigating Officer, copied to the District Director.
- Tone: Respectful acknowledgment of the 483 observations and thanks to the team.
- Commitment: Bold, unwavering commitment from senior management to rectify all issues and ensure systemic cGMP compliance.
- Summary: A high-level overview of the most critical actions already completed (Day 0–15).
- Signatory: Senior Executive (VP of QA or Plant Head).
II. Body of Response (Detailed Tabulated Action Plan)
The most effective method is a tabular format that addresses each observation systematically.
| FDA Observation # | FDA Text (Copy Exact) | Background/Factual Clarification | Root Cause (Systemic Finding) | Immediate Correction (Completed) | Corrective Action (CA) & Preventive Action (PA) | Responsible Party | Completion Date (Specific Date) | Objective Evidence (Attachment Ref.) |
| 1. | (e.g., Procedures are not followed for cleaning the mixer) | The procedure lacked clarity regarding use of clean room gloves during transfer. | Inadequate design control of SOPs (systemic issue in Change Control QMS). | Retraining of 4 operators was completed on 10/18/2025. | Revise SOP XX-007 to include visual aids for gowning/transfer (PA). Initiate QMS project to audit 10 critical SOPs for clarity (PA). | Head of Manufacturing & QA Manager | 12/30/2025 | Attachment C: Training Logs; Attachment D: Change Control CC-2025-502. |
| 2. | (Repeat Finding Example) | … | Failure of previous CAPA to correctly identify systemic training gap. | … | CAPA on CAPA: Initiate external QMS audit of the training system by 1/15/2026. Revise training matrix site-wide. | VP of Quality | 03/01/2026 | … |
III. Attachments (The Objective Evidence)
- All revised procedures (SOPs).
- Signed training records/log-in data for electronic training.
- Change Control/Deviation reports related to the observations.
- Validation protocols and summaries (e.g., if a new software audit trail was validated).
- Product Impact Assessment Report summary.
Future-Proofing Tactics: Sustainable Compliance for Tomorrow
The goal of the 483 response should be to not receive another. This requires a cultural and technological investment.
Culture Improvement: The Compliance Ownership Model
- Decentralized Quality: Move from a centralized QA “police” model to one where quality ownership is embedded within Manufacturing and Engineering. Compliance should be seen as a strategic competitive advantage, not a regulatory burden.
- Senior Management Walk-Throughs: Encourage frequent, unannounced walk-throughs by senior leadership (Plant Head, VP of Ops) to identify subtle compliance drift before it becomes a 483 observation.
Digital Documentation and ALCOA++ Systems
- Validation of Cloud/Digital QMS: Ensure your electronic Quality Management System (eQMS) is fully validated and configured to prevent the possibility of data integrity issues. Implement systems that automatically enforce ALCOA++ (e.g., mandatory audit trails, sequential data capture, and restricted access).
- Automated Trending: Implement automated quality metrics trending (deviations, CAPAs, OOS results) to detect negative trends early. The FDA expects companies to use these metrics to drive continuous improvement, not just for reporting.
Myth Busting & FAQs
1. Myth: Arguing against an observation will get it removed.
Reality: Almost never. The 483 is the investigator’s judgment. Arguing is perceived as defensive and uncooperative. Acknowledge the observation, and then use the Background/Factual Clarification section of your response to respectfully clarify any factual misunderstandings that led to the observation. Focus on what you did or will do, not why the observation was wrong.
2. Myth: The 15-day response timeline is optional.
Reality: While not a legal requirement, it is a firm policy expectation. Submitting a response after 15 days is often viewed as non-cooperation and is frequently cited as a reason to issue a Warning Letter without reviewing the response content.
3. Myth: The CAPA must be fully implemented within 15 days.
Reality: False. The plan must be submitted within 15 days. It must be a detailed, time-bound plan with realistic dates. Long-term CAPAs (e.g., facility upgrades) are acceptable, provided you commit to regular updates.
4. Myth: We only need to fix the specific product/area cited.
Reality: False. This is a common pitfall leading to repeat findings. The FDA expects a systemic investigation—if a procedure failed in one area, the regulatory expectation is to determine the vulnerability in the underlying QMS element (e.g., Change Control or Training) and fix it across all affected departments/sites.
5. Myth: External consultants should write the entire response.
Reality: Consultants can advise and structure the response, but the content, root cause, and commitment must come from the site’s QA, Operations, and Senior Management. The FDA wants to see your company’s ownership and quality culture reflected in the narrative.
Conclusion: The Immediate Action Checklist for Decision-Makers
The 483 response is your company’s golden opportunity to demonstrate mastery of cGMP and proactive compliance. View it not as a penalty, but as a framework for systemic improvement.
| Priority | Action Item | Accountability (Example) |
| Immediate (Day 0–3) | Assign the Senior Executive Response Team Leader. | VP of Quality |
| High (Day 3–7) | Initiate formal Root Cause Analysis (Fishbone/FMEA) for all observations. | QA Manager |
| High (Day 7–10) | Complete Product Impact Assessment (SISPQ) for all affected lots. Implement Quality Holds. | Head of QC/Batch Release |
| Critical (Day 12–15) | Review and sign the Cover Letter (Commitment) and the final response table. Ensure the 5 Cs are met. | CEO/President/Plant Head |
| Ongoing (Post-Submission) | Schedule the first CAPA status update (within 30 days) and define effectiveness checks (e.g., 6–12 month lookback). | Regulatory Affairs/CAPA Owner |
Ready to transform your FDA 483 response from a regulatory burden into a strategic advantage? Download our exclusive ‘2025 FDA 483 Response Checklist & CAPA Template’ now and ensure your next inspection is a testament to your unyielding commitment to quality.
References
- U.S. Food and Drug Administration (FDA). Inspectional Observations and Citations. [https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspectional-observations-and-citations]
- U.S. Food and Drug Administration (FDA). Frequently Requested or Proactively Posted Compliance Records. [https://www.fda.gov/drugs/cder-foia-electronic-reading-room/frequently-requested-or-proactively-posted-compliance-records]
- The FDA Group (via IntuitionLabs). FDA 483 Response: A Guide to Best Practices & Compliance.[https://intuitionlabs.ai/articles/fda-483-response-best-practices]
- Leucine.io. Your FDA 483 Response Guide: 15-Day Action Plan. [https://www.leucine.io/fda-tracker-insights/fda-483-response]
- Clinical Leader / Outsourced Pharma. 5 Cs For Responding To FDA-483s — Strategies For Effective Compliance And Resolution. [https://www.clinicalleader.com/doc/cs-for-responding-to-fda-s-strategies-for-effective-compliance-and-resolution-0001]
- Pharmaceutical Technology (PharmTech). PDA 2025: FDA Experts’ Proactive Strategies for Mastering 483 Responses.[https://www.pharmtech.com/view/pda-2025-fda-experts-proactive-strategies-for-mastering-483-responses]
- Greenlight Guru. The Definitive Guide to Responding to FDA 483 and Warning Letters. [https://www.greenlight.guru/hubfs/Sales_Material/gg_Guide_to_FDA_483_Warning_Letters.pdf]
- Redica Systems. FDA 483 Database / The Ultimate Guide to Form FDA 483s.[https://redica.com/lp-fda-483-database/]
- NSF International. Ten Steps to an Effective Medical Devices FDA 483 Response.[https://www.nsf.org/knowledge-library/ten-steps-effective-medical-devices-fda-483-responses]
- Kapstone Medical. What is an FDA 483, and How Do You Respond? [ https://www.kapstonemedical.com/resource-center/blog/what-is-an-fda-483]
- Scilife.io. How to respond to FDA warning letters: Best practices and tips. [https://www.scilife.io/blog/fda-warning-letters-483-observations-response]
- American Pharmaceutical Review. FDA 483s and Non-Compliance in Pharma.[https://www.americanpharmaceuticalreview.com/589645-FDA-483s-and-Non-Compliance-in-Pharma/]



: A Potential Breakthrough in Diabetes Treatment")