The recent FDA approval of WAYRILZ (rilzabrutinib) marks a significant advancement for adults with persistent or chronic immune thrombocytopenia (ITP) who have had an insufficient response to previous treatments. Developed by Genzyme, a Sanofi company, this novel Bruton’s tyrosine kinase (BTK) inhibitor demonstrated considerable efficacy in the pivotal LUNA-3 clinical trial, offering a new therapeutic pathway for a challenging condition[1, 3].
However, a drug’s journey doesn’t end at its approval date. In fact, for many novel therapies, the approval is the beginning of a new phase of rigorous evaluation. As part of its August 29, 2025, approval of WAYRILZ, the FDA mandated a series of postmarketing requirement (PMR) studies under section 505(o) of the Federal Food, Drug, and Cosmetic Act (FDCA) [2, 4]. These studies are not a sign of concern but a proactive measure to build a more comprehensive understanding of the drug’s long-term safety and efficacy in real-world patient populations [4].
This article provides a deep dive into the four specific PMRs for WAYRILZ, exploring the scientific questions they aim to answer, why they are clinically necessary, and what the findings will mean for the future of ITP treatment.
Approval for WAYRILZ?
WAYRILZ’s approval was based on the strength of the LUNA-3 study, a randomized, double-blind, placebo-controlled trial that evaluated its efficacy in 202 adult patients with ITP. The trial’s primary endpoint was durable platelet response, a stringent measure defined as achieving a weekly platelet count ≥50×109/L for at least two-thirds of the final 12 weeks of the 24-week study period, without the need for rescue therapy [1, 5].
The results were statistically significant and clinically meaningful:
- WAYRILZ Group: 23.3% of patients (31 out of 133) achieved a durable platelet response.
- Placebo Group: 0% of patients (0 out of 69) achieved this endpoint.
Furthermore, patients treated with WAYRILZ required rescue medication less frequently than those on placebo (33% vs. 58%, respectively) and had a median time to first platelet response of just 36 days. This data establishes WAYRILZ as an effective new oral therapy, taken at a dose of 400 mg twice daily, for a patient population with significant unmet needs.
Why FDA Approval is Just the Beginning?
While pivotal trials like LUNA-3 are essential for establishing initial safety and efficacy, they are conducted under highly controlled conditions and often exclude patients with certain comorbidities or characteristics (e.g., pregnant women, individuals with severe organ impairment) [6]. The FDA’s authority to require PMRs under FDCA section 505(o)(3) is a critical tool for bridging the gap between clinical trial data and real-world clinical practice [4].
These studies are mandated when the FDA determines that an analysis of spontaneous adverse event reporting alone is not sufficient to assess a known serious risk or a signal of a serious risk associated with a drug [4]. For WAYRILZ, the FDA identified four specific areas where more data is needed to refine the drug’s safety profile and provide more definitive guidance to clinicians and patients [2].
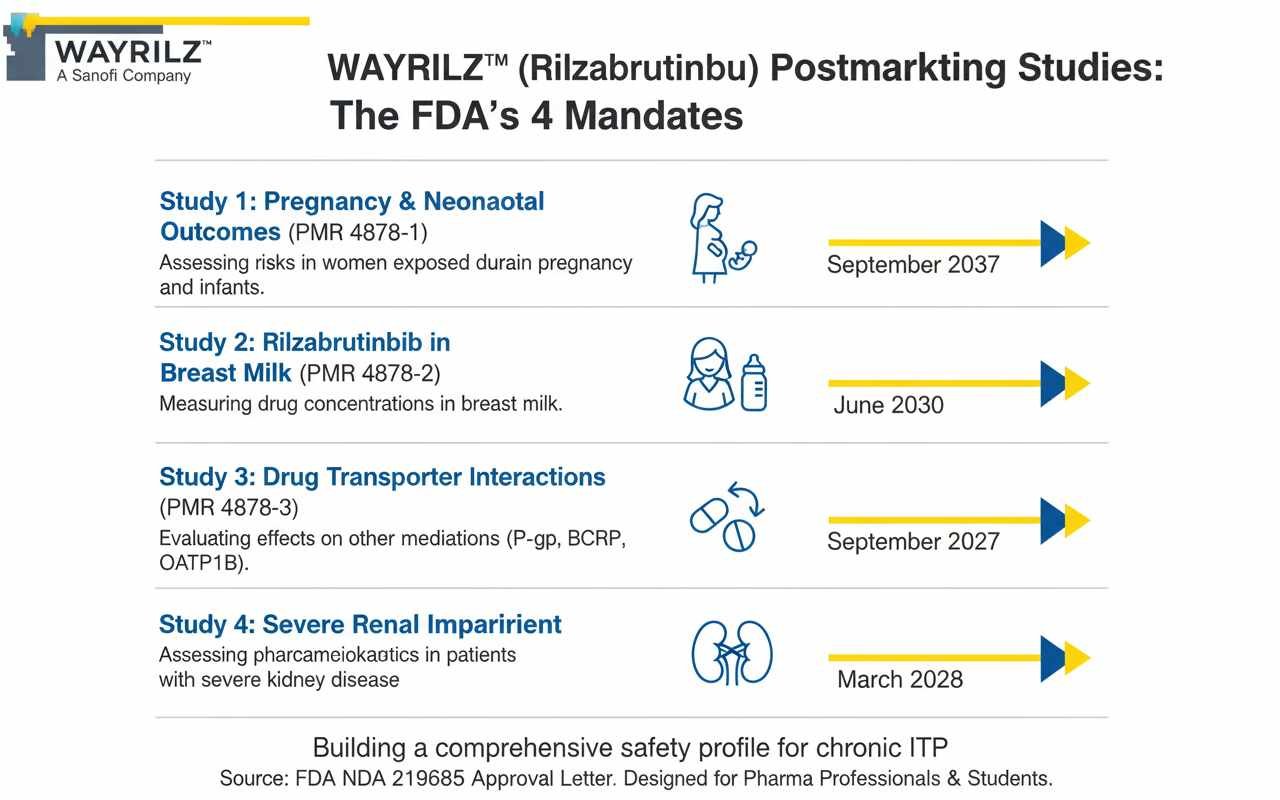
The 4 Mandated Studies for Rilzabrutinib
The FDA has required Genzyme to conduct two observational studies and two clinical trials to address specific knowledge gaps in the current WAYRILZ label.
| PMR Number | Study Type | Objective | Key Knowledge Gap Addressed | Final Report Due | ||
| 4878-1 | Observational | Assess risks in women exposed to WAYRILZ during pregnancy and their infants. | No human pregnancy data is currently available; the label’s warning is based on animal studies. | September 2037 | ||
| 4878-2 | Clinical (Milk-Only) | Measure rilzabrutinib concentrations in the breast milk of lactating women. | It is unknown if rilzabrutinib passes into human breast milk. | June 2030 | ||
| 4878-3 | Clinical Trial | Evaluate the effect of WAYRILZ on substrates of P-gp, BCRP, OATP1B1, and OATP1B3 transporters. | In vitro data suggests inhibitory potential, but the clinical impact on other drugs has not been established. | September 2027 | ||
| 4878-4 | Clinical Trial | Assess WAYRILZ pharmacokinetics in patients with severe renal impairment. | Patients with severe renal impairment (eGFR <30 mL/min) were not studied in pre-approval trials. | March 2028 |
Study 1: Assessing Pregnancy and Neonatal Outcomes (PMR 4878-1)
- The Question: How does WAYRILZ exposure during pregnancy affect the mother, fetus, and infant?
- Why It’s Needed: The current prescribing information for WAYRILZ states that it “may cause fetal harm when administered to a pregnant woman” [1]. This warning is based on preliminary animal reproduction studies, which found adverse skeletal and visceral findings in rat fetuses at maternally toxic doses [1]. However, the label explicitly states, “There are no available clinical data on the use of WAYRILZ during pregnancy” [1]. This study, a worldwide descriptive analysis of prospective and retrospective data, will provide the first human data to quantify the actual risk of maternal complications and adverse fetal or infant outcomes, with infant follow-up through at least the first year of life [2].
- Timeline: This is a long-term observational study, reflecting the time needed to collect a sufficient number of exposures. The final protocol is due by December 2026, with the final report submission slated for September 2037 [2].
Study 2: Quantifying Rilzabrutinib in Breast Milk (PMR 4878-2)
- The Question: Does rilzabrutinib pass into breast milk, and if so, at what concentration?[2]
- Why It’s Needed: Similar to pregnancy, there is a complete lack of data on the drug’s presence in human milk. The label states, “There are no data on the presence of rilzabrutinib or its metabolites in either human or animal milk” [1]. This absence of information necessitates a conservative recommendation: advise lactating women not to breastfeed during treatment and for one week after the final dose [1]. This “milk-only” lactation study will use a validated assay to measure rilzabrutinib concentrations directly in breast milk, providing the scientific basis for more informed guidance [2].
- Timeline: The final protocol is due by December 2026, with the final report expected by June 2030 [2].
Study 3: Investigating Drug Transporter Interactions (PMR 4878-3)
- The Question: Does co-administration of WAYRILZ alter the exposure of other drugs that are substrates of key transporters like P-glycoprotein (P-gp), BCRP, and OATP1B?
- Why It’s Needed: Rilzabrutinib is an inhibitor of these transporters in vitro (in a lab setting). Many common medications—including certain statins, anticoagulants, and chemotherapies—rely on these transporters for proper absorption and elimination. While the label advises monitoring for adverse reactions when WAYRILZ is used with these substrates, it also notes that “the effect of concomitant use…has not been established in clinical studies”. This clinical drug interaction study in healthy volunteers will quantify the magnitude of these potential interactions, informing whether more specific dosing adjustments or contraindications are necessary.
- Timeline: The final protocol is due by June 2026, with the final report scheduled for September 2027.
Study 4: Clarifying Dosing in Severe Renal Impairment (PMR 4878-4)
- The Question: How does severe renal impairment (eGFR <30 mL/min) affect the body’s exposure to WAYRILZ, and is a dose adjustment required?
- Why It’s Needed: The pre-approval pharmacokinetic studies for WAYRILZ did not include patients with severe renal impairment. Due to this lack of data, the current label recommends avoiding its use in this population. This dedicated clinical trial will compare the pharmacokinetics of rilzabrutinib in subjects with severe renal impairment to those with normal renal function. The results will determine if the drug can be used safely in this population, potentially with a modified dose, thereby expanding access for patients with both chronic ITP and significant kidney disease.
- Timeline: The final protocol is due by June 2026, with the final report expected by March 2028.
What This Means for Clinicians and Patients?
The FDA’s mandate for these four postmarketing studies underscores a commitment to evidence-based medicine and patient safety. While WAYRILZ has already demonstrated a favorable risk-benefit profile for its approved indication, these studies will systematically address the remaining uncertainties.
For clinicians and patients today, the message is clear: adhere to the guidance in the current, approved prescribing information. This includes:
- Verifying the pregnancy status of females of reproductive potential before starting treatment and advising the use of effective contraception.
- Advising patients not to breastfeed during and for one week after treatment.
- Avoiding use in patients with severe renal or moderate-to-severe hepatic impairment.
- Carefully monitoring patients taking concurrent medications that are substrates of P-gp, BCRP, or OATP1B.
Over the next decade, the data generated from these PMRs will be submitted to the FDA, likely resulting in updates to the WAYRILZ label. This will provide a more nuanced and complete safety profile, empowering healthcare providers to make more personalized and confident treatment decisions for their patients with chronic ITP. The journey of rilzabrutinib is far from over; it is evolving toward a more comprehensive understanding that will ultimately benefit the entire ITP community.
FAQs
What is a postmarketing requirement (PMR) study?
A PMR is a study or clinical trial that the FDA requires a pharmaceutical company to conduct after a drug has been approved. These studies are used to gather additional information about a drug’s safety, efficacy, or optimal use in populations not fully assessed in pre-approval trials.
Does the fact that WAYRILZ has PMRs mean it’s unsafe?
No. Mandating PMRs is a standard part of the regulatory process for many new drugs. It does not imply that the drug is unsafe for its approved indication. It means the FDA has identified specific areas where more data is needed to create a more complete long-term safety profile and refine prescribing information.
Can I take WAYRILZ if I am pregnant or breastfeeding?
Based on the current FDA-approved label, you should not. The label states WAYRILZ may cause fetal harm and recommends females of reproductive potential use effective contraception. It also advises women not to breastfeed during treatment and for one week after the last dose because it is unknown if the drug passes into breast milk. The ongoing PMRs will provide more definitive data to guide these recommendations in the future.
Why can’t patients with severe kidney disease take WAYRILZ?
The current label advises avoiding WAYRILZ in patients with severe renal impairment because this population was not included in the clinical trials conducted for the drug’s approval. Therefore, its safety and appropriate dosage in these patients are unknown. A required postmarketing clinical trial (PMR 4878-4) is underway to study this exact issue and determine if the drug can be used safely in this group.
What is the LUNA-3 study?
The LUNA-3 study was the primary clinical trial that supported the FDA approval of WAYRILZ for chronic ITP. It was a randomized, double-blind, placebo-controlled study that proved WAYRILZ was significantly more effective than a placebo at producing a durable increase in platelet counts in adult patients who had failed prior treatments.
How long will it take to get results from these new studies?
These studies are long-term commitments. The final report deadlines range from September 2027 for the drug interaction study to as far out as September 2037 for the pregnancy outcomes study. Interim reports will be submitted to the FDA periodically before these final deadlines.
Summary
The FDA’s mandate for four postmarketing studies on WAYRILZ™ underscores a commitment to long-term patient safety. While the LUNA-3 trial established rilzabrutinib’s efficacy for chronic ITP, these required studies will provide critical real-world data on its use during pregnancy, lactation, and in patients with severe renal impairment. They will also clarify potential drug interactions. Over the next decade, these findings will refine the drug’s safety profile, enabling clinicians to make more informed and personalized treatment decisions for the ITP community.
References
- WAYRILZ (rilzabrutinib) Prescribing Information. U.S. Food and Drug Administration. August 2025. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219685s000lbl.pdf (Accessed: 11 Sep 2025).
- NDA 219685 Approval Letter. U.S. Food and Drug Administration. 29 Aug 2025. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2025/219685s000ltr.pdf (Accessed: 11 Sep 2025).
- Immune Thrombocytopenia. National Organization for Rare Disorders (NORD). 2023. Available at: https://rarediseases.org/rare-diseases/immune-thrombocytopenia/ (Accessed: 11 Sep 2025).
- Guidance for Industry: Postmarketing Studies and Clinical Trials — Implementation of Section 505(o)(3) of the Federal Food, Drug, and Cosmetic Act. U.S. Food and Drug Administration. October 2019. Available at: https://www.fda.gov/media/71447/download (Accessed: 11 Sep 2025).
- A Study to Test Efficacy and Safety of Rilzabrutinib in Adult and Adolescent Participants With Persistent or Chronic Immune Thrombocytopenia (ITP) (LUNA 3). ClinicalTrials.gov. Identifier: NCT04562766. Available at: https://clinicaltrials.gov/study/NCT04562766 (Accessed: 11 Sep 2025).
- Enhancing the Diversity of Clinical Trial Populations — Eligibility Criteria, Enrollment Practices, and Trial Designs Guidance for Industry. U.S. Food and Drug Administration. November 2020. Available at: https://www.fda.gov/media/127712/download (Accessed: 11 Sep 2025).
- Role of Drug Transporters in Drug Disposition, Drug-Drug Interactions and Pharmacogenetics. S. K. Gupta. Scientia Pharmaceutica. 2010. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3102553/ (Accessed: 11 Sep 2025).
- Guidance for Industry: Pharmacokinetics in Patients With Impaired Renal Function — Study Design, Data Analysis, and Impact on Dosing and Labeling. U.S. Food and Drug Administration. March 2020. Available at: https://www.fda.gov/media/71355/download (Accessed: 11 Sep 2025).

: MOA, Approvals & India")

